Getting Started with PLAID
BigOmics Analytics
Source:vignettes/01_plaid-vignette.Rmd
01_plaid-vignette.RmdIntroduction
PLAID (Pathway Level Average Intensity Detection) is a novel, ultrafast and memory optimized gene set enrichment scoring algorithm. PLAID demonstrates accurate gene set scoring and outperforms all currently available gene set scoring methods in large bulk and single-cell RNA-seq datasets.
Motivation
In recent years, computational methods have emerged that calculate enrichment of gene signatures within individual samples, rather than across pooled samples. These signatures offer critical insights into the coordinated activity of functionally related genes, proteins or metabolites, enabling identification of unique molecular profiles based on gene set and pathway activity in individual cells and patients. This strategy is pivotal for patient stratification and advancement of personalized medicine. However, the rise of large-scale datasets, including single-cell profiles and population biobanks, has exposed significant computational inefficiencies in existing methods. Current methods often demand excessive runtime and memory resources, becoming impractical for large datasets. Overcoming these limitations is a focus of current efforts by bioinformatics teams in academia and the pharmaceutical industry, as essential to support basic and clinical biomedical research.
Example: Single-cell RNA-seq hallmark scoring
Preparing data
For this vignette, our package includes a small subset of the the pmbc3k single-cell dataset of just 50 cells. Please install the Seurat and SeuratData packages if you want to run this vignette against the full dataset.
library("plaid")
#> Warning: replacing previous import 'matrixStats::colRanks' by
#> 'sparseMatrixStats::colRanks' when loading 'plaid'
load(system.file("extdata", "pbmc3k-50cells.rda", package = "plaid"),verbose=TRUE)
#> Loading objects:
#> X
#> celltype
dim(X)
#> [1] 7728 50Note that X is the normalized expression matrix from the Seurat object, not the raw counts matrix. We recommend to run plaid on the log transformed expression matrix, not on the counts, as the average in the logarithmic space is more robust and is in concordance to calculating the geometric mean.
It is not necessary to normalize your expression matrix before running plaid because plaid normalizes the enrichment scores afterwards. However, again, log transformation is recommended.
It is recommended to keep the expression matrix sparse as much as possible because plaid extensively take advantage of sparse matrix computations. But even for dense matrices plaid is fast.
Preparing gene sets
For convenience we have included the 50 Hallmark genesets in our package. But we encourage you to download larger geneset collections as plaid’s speed advantage will be more apparent for larger datasets and large geneset collections.
Plaid needs the gene sets as sparse matrix. If you have your collection of gene sets a a list, we need first to convert the gmt list to matrix format.
hallmarks <- system.file("extdata", "hallmarks.gmt", package = "plaid")
gmt <- read.gmt(hallmarks)
matG <- gmt2mat(gmt)
dim(matG)
#> [1] 4386 50If you have your own gene sets stored as gmt files, you can
conveniently use the included read.gmt() function to read
the gmt file.
Calculating the score
The main function to run plaid is plaid(). We run plaid
on our expression matrix X and gene set matrix
matG.
The resulting matrix gsetX contains the single-sample
enrichment scores for the specified gene sets and samples.
Notice that by default plaid performs median normalization of the final results. That also means that it is not necessary to normalize your expression matrix before running plaid. However, generally, log transformation is recommended.
Plaid can also be run on the ranked matrix, we will see later that this corresponds to the singscore (Fouratan et al., 2018). Or plaid could be run on the (non-logarithmic) counts which can be used to calculate the scSE score (Pont et al., 2019).
Very large matrices
Plaid is fast and memory efficient because it uses very efficient
sparse matrix computation in the back. For very large X,
plaid uses chunked computation by splitting the matrix in chunks to
avoid index overflow. Should you encounter errors, please compute your
dataset by subsetting manually the expression matrix and/or gene
sets.
Although X and matG are generally very
sparse, be aware that the result matrix gsetX generally is
dense and therefore can become very large. If you would want to compute
the score of 10.000 gene sets on a million of cells this would create a
large 10.000 x 1.000.000 dense matrix which requires about 75GB of
memory.
Differential expression testing using dualGSEA
Once we have the gene sets scores we can use these scores for
statistical analysis. We could compute the differential gene set
expression between two groups using a general t-test or limma directly
on the score matrix gsetX.
Another way to test whether a gene set is statistically significant
would be to test whether the fold-change of the genes in the gene sets
are statistically different than zero. That is, we can perform a one
sample z-test on the logFC of the genes of each gene sets and test
whether they are significantly different from zero. The logFC is
computed from the original (log) expression matrix X and
group vector y.
The function dualGSEA() does both tests: the one-sample
z-test on the logFC and the two-group t-test on the gene set matrix
gsetX. Dual testing has been suggested by Bull et al. (Sci
Rep., 2024)
y <- 1*(celltype == "B")
res <- dualGSEA(X, y, G=matG)
#> FC testing using rankcor
#> single-sample testing using plaidThe top significant genesets can be shown with
res <- res[order(res[,"p.dual"]),]
head(res)
#> gsetFC size p.fc p.ss
#> HALLMARK_INTERFERON_GAMMA_RESPONSE 0.09923298 155 0.01254942 1.067381e-05
#> HALLMARK_P53_PATHWAY -0.04702449 130 0.01642868 2.900893e-04
#> HALLMARK_ALLOGRAFT_REJECTION 0.08322962 134 0.28923587 1.311508e-05
#> HALLMARK_INTERFERON_ALPHA_RESPONSE 0.09705679 78 0.01820425 9.406415e-03
#> HALLMARK_PEROXISOME 0.04304130 57 0.02263533 3.818109e-02
#> HALLMARK_HYPOXIA -0.03466136 96 0.02063956 5.356212e-02
#> p.dual q.dual
#> HALLMARK_INTERFERON_GAMMA_RESPONSE 2.224153e-06 0.0001112076
#> HALLMARK_P53_PATHWAY 4.043737e-05 0.0010109343
#> HALLMARK_ALLOGRAFT_REJECTION 3.819973e-04 0.0063666222
#> HALLMARK_INTERFERON_ALPHA_RESPONSE 8.428564e-04 0.0105357051
#> HALLMARK_PEROXISOME 3.805555e-03 0.0380555487
#> HALLMARK_HYPOXIA 4.906745e-03 0.0408895398The column gsetFC corresponds to the difference in gene
set score and also corresponds to the average foldchange of the genes in
the gene set. The column ‘p.fc corresponds to the test on the preranked
logFC, the column ’p.ss’ corresponds to the two-group t-test on the
geneset scores gsetX. The two p-values are then combined
using Stouffer’s method in the column ‘p.dual’ and adjusted for multiple
testing in column q.dual.
The left figure below plots the fold-change enrichment
p.fc vs. single-sample enrichment p.ss. The
right figure shows the volcano plot p.dual
vs. gsetFC:
fc <- res[,"gsetFC"]
pv <- res[,"p.dual"]
p1 <- res[,"p.fc"]
p2 <- res[,"p.ss"]
ii <- head(order(pv))
par(mfrow=c(1,2))
plot( -log10(p1), -log10(p2),
xlab="FC enrichment (-log10p)",
ylab="single-sample enrichment (-log10p)", pch=19)
text( -log10(p1[ii]), -log10(p2[ii]), rownames(res)[ii],pos=2)
plot( fc, -log10(pv), xlab="gsetFC", ylab="-log10p", pch=19)
abline(h=0, v=0, lty=2)
text( fc[ii], -log10(pv[ii]), rownames(res)[ii],pos=2)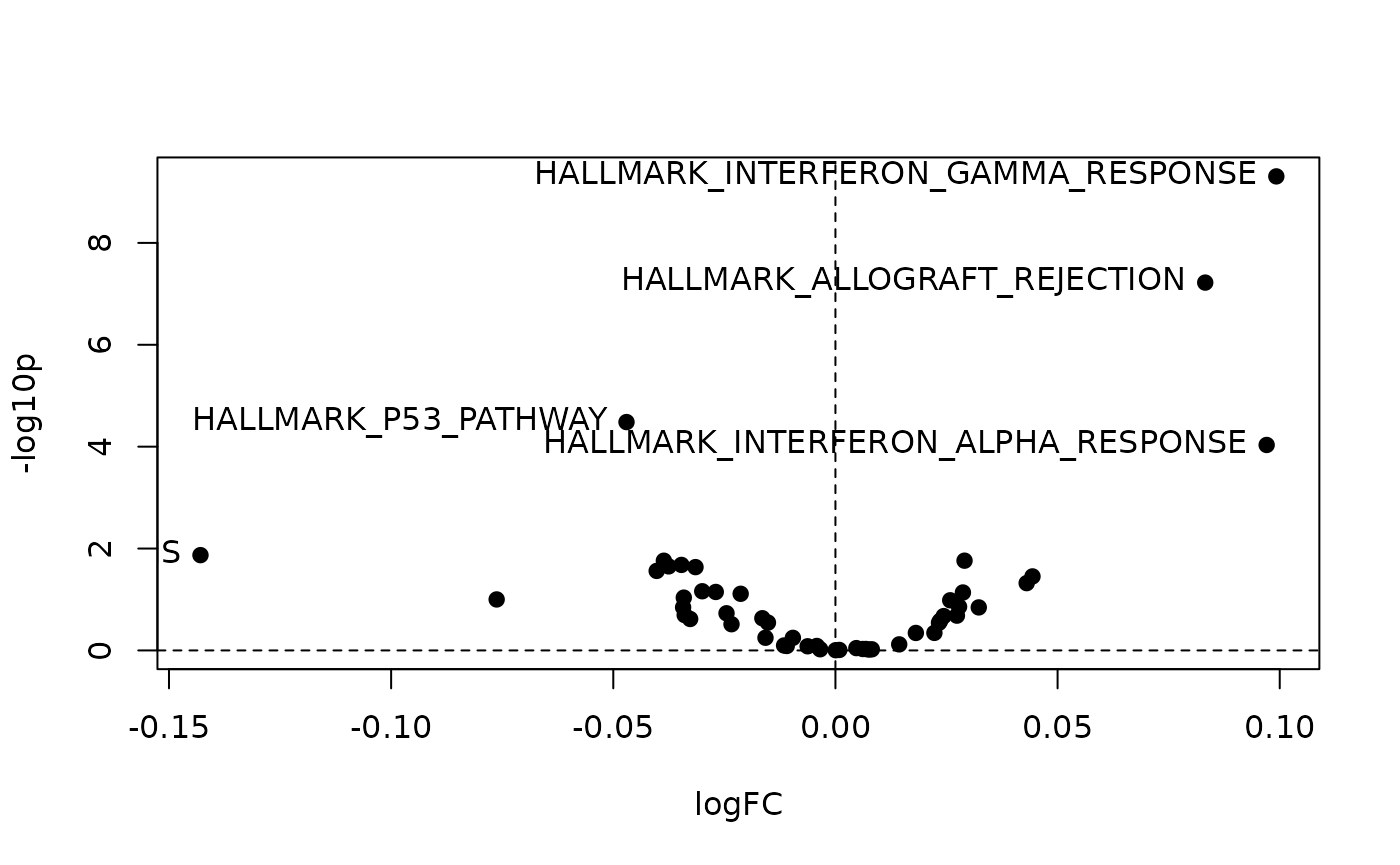
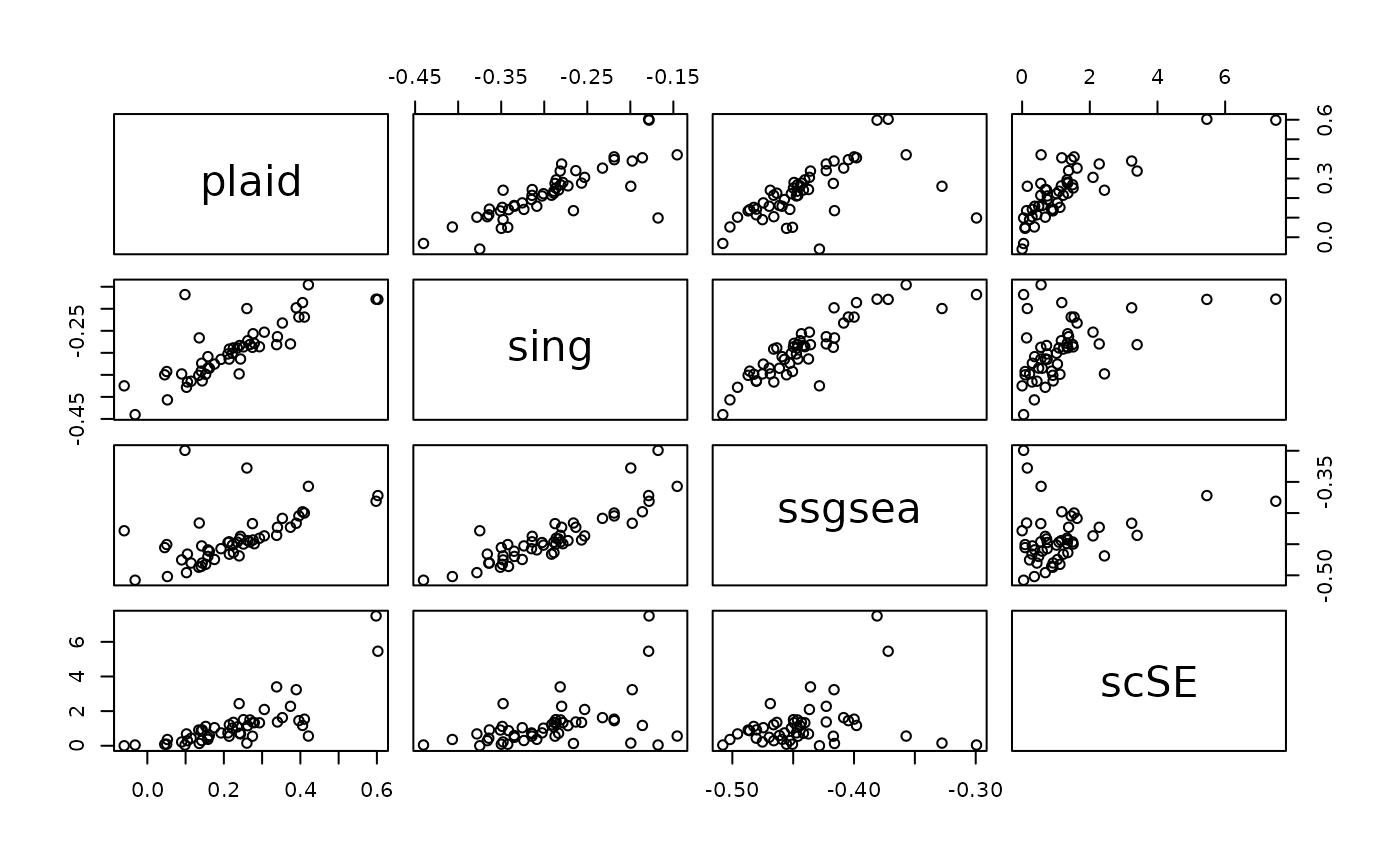
Replicating ssGSEA, singscore and scSE
Plaid can be used to replicate other enrichment score such as ssGSEA, GSVA, AUCell, Singscore, scSE and UCell. But using plaid, the computation is much faster than the original code.
Replicating singscore
Computing the singscore requires to compute the ranks of the expression matrix. We have wrapped this in a single convenience function:
sing <- replaid.sing(X, matG)We have extensively compared the results of replaid.sing
and from the original singscore R package and we showed
identical result in the score, logFC and p-values.
Replicating ssGSEA
Plaid can also be used to compute the ssGSEA score (Barbie et al., 2009). Using plaid, we can calculate the score upto 100x faster. We have wrapped this in a single convenience function:
ssgsea <- replaid.ssgsea(X, matG, alpha=0)We have extensively compared the results of
replaid.ssgsea() and from the original GSVA R package. Note the
rank weight parameter alpha. For alpha=0 we obtained
identical result for the score, logFC and p-values. For non-zero values
for alpha the results are close but not exactly the same. The default
value in the original publication and in GSVA is
alpha=0.25.
Replicating the scSE score
Single-cell Signature Explorer (scSE) is a fast enrichment algorithm implemented in GO. Computing the scSE score requires running plaid on the linear (not logarithmic) score and perform additional normalization by the total UMI per sample. We have wrapped this in a single convenience function:
scse <- replaid.scse(X, matG, removeLog2=TRUE, scoreMean=FALSE)
#> [replaid.scse] Converting data to linear scale (removing log2)...To replicate the original “sum-of-UMI” scSE score, set
removeLog2=TRUE and scoreMean=FALSE. scSE and
plaid scores become more similar for removeLog2=FALSE and
scoreMean=TRUE.
We have extensively compared the results from
replaid.scse and from the original scSE (implemented in GO
lang) and we showed almost identical results in the score, logFC and
p-values.
Session info
sessionInfo()
#> R version 4.3.3 (2024-02-29)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.12.0
#> LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: Europe/Zurich
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] plaid_0.99.12 BiocStyle_2.30.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.1 dplyr_1.1.4
#> [3] blob_1.2.4 Biostrings_2.70.3
#> [5] bitops_1.0-9 fastmap_1.2.0
#> [7] RCurl_1.98-1.17 digest_0.6.38
#> [9] sparsesvd_0.2-3 lifecycle_1.0.4
#> [11] KEGGREST_1.42.0 RSQLite_2.4.4
#> [13] magrittr_2.0.4 compiler_4.3.3
#> [15] rlang_1.1.6 sass_0.4.10
#> [17] tools_4.3.3 yaml_2.3.10
#> [19] knitr_1.50 S4Arrays_1.2.1
#> [21] htmlwidgets_1.6.4 ontologyIndex_2.12
#> [23] bit_4.6.0 DelayedArray_0.28.0
#> [25] plyr_1.8.9 abind_1.4-8
#> [27] purrr_1.2.0 BiocGenerics_0.48.1
#> [29] desc_1.4.3 grid_4.3.3
#> [31] stats4_4.3.3 qlcMatrix_0.9.9
#> [33] docopt_0.7.2 SummarizedExperiment_1.32.0
#> [35] cli_3.6.5 rmarkdown_2.30
#> [37] crayon_1.5.3 ragg_1.5.0
#> [39] generics_0.1.4 RcppParallel_5.1.11-1
#> [41] httr_1.4.7 DBI_1.2.3
#> [43] cachem_1.1.0 zlibbioc_1.48.2
#> [45] parallel_4.3.3 AnnotationDbi_1.64.1
#> [47] BiocManager_1.30.27 XVector_0.42.0
#> [49] matrixStats_1.5.0 vctrs_0.6.5
#> [51] Matrix_1.6-5 jsonlite_2.0.0
#> [53] slam_0.1-55 bookdown_0.45
#> [55] IRanges_2.36.0 S4Vectors_0.40.2
#> [57] bit64_4.6.0-1 systemfonts_1.3.1
#> [59] jquerylib_0.1.4 tidyr_1.3.1
#> [61] glue_1.8.0 pkgdown_2.2.0
#> [63] GenomeInfoDb_1.38.8 BiocIO_1.12.0
#> [65] GenomicRanges_1.54.1 BiocSet_1.16.1
#> [67] tibble_3.3.0 pillar_1.11.1
#> [69] htmltools_0.5.8.1 GenomeInfoDbData_1.2.11
#> [71] R6_2.6.1 zigg_0.0.2
#> [73] textshaping_1.0.4 sparseMatrixStats_1.14.0
#> [75] evaluate_1.0.5 Biobase_2.62.0
#> [77] lattice_0.22-7 png_0.1-8
#> [79] Rfast_2.1.5.2 memoise_2.0.1
#> [81] bslib_0.9.0 Rcpp_1.1.0
#> [83] SparseArray_1.2.4 xfun_0.54
#> [85] fs_1.6.6 MatrixGenerics_1.14.0
#> [87] pkgconfig_2.0.3